
膜タンパク
膜タンパクのαヘリックス構造は、リン脂質膜に膜タンパクを固定するアンカーの役割がある。G蛋白は7つのαヘリックス構造を持つ。
Gs蛋白はグルカゴン、TSH、PTHの受容体に存在し、アデニル酸シクラーゼを活性化する。それによりATPがcAMPに変換され、cAMPはプロテインキナーゼAを活性化する。それによりDNAに結合する蛋白がリン酸化される。
ホルモンレセプター
脂溶性ホルモンの場合は、ホルモンが膜を通過し、細胞内レセプターと結合する。このレセプターはジンクフィンガーを持っており、直接ターゲットとなるDNA配列に結合し、遺伝子発現を調整する。
このようなDNA結合蛋白には転写因子(Myc, CREB)、ステロイド、サイロイドホルモン、脂溶性ビタミン(ビタミンD、ビタミンA)、DNA転写、複製蛋白がある。
水溶性ホルモンの場合は、膜貫通性レセプターに結合し、レセプターからセカンドメッセンジャーが放出される。それによりDNA配列へ結合する別の蛋白が活性化される。
成長因子のシグナル伝達経路
- MAPキナーゼ経路
- PI3K/Akt/mTOR経路:抗apotosis、細胞増殖、血管新生
- イノシトール リン脂質経路
- cAMP経路
- JAK/STAT経路:成長ホルモン
競合的阻害
最大反応速度は下げないが、被反応物が低量時に反応速度が下がる。
小胞体
滑面小胞体 smooth endoplasmic reticulumは有害物の解毒、炭水化物代謝、脂質合成、脂溶性ホルモンの生成、分泌に関連。例:ステロイド、甲状腺ホルモン
粗面小胞体 rough endoplasmic reticulumはリボソームと結合しており、蛋白の細胞膜、細胞外への輸出、ポリペプチドホルモンに関連。例:ADH、インスリン、ガストリン、プロラクチン、エピネフリン、成長ホルモン等
ゴルジ体
蛋白の修飾、組み換え、輸送
リソソーム
細胞残渣、病原体の消化
プロテアソーム
ユビキチン化された蛋白の分解
ミトコンドリア
TCAサイクル、脂肪酸の酸化。ATP産生。アポトーシス
ペルオキシソーム
長分枝脂肪酸(VLCFA)の酸化。ハイドロゲンペルオキシドの分解
代謝経路のまとめ
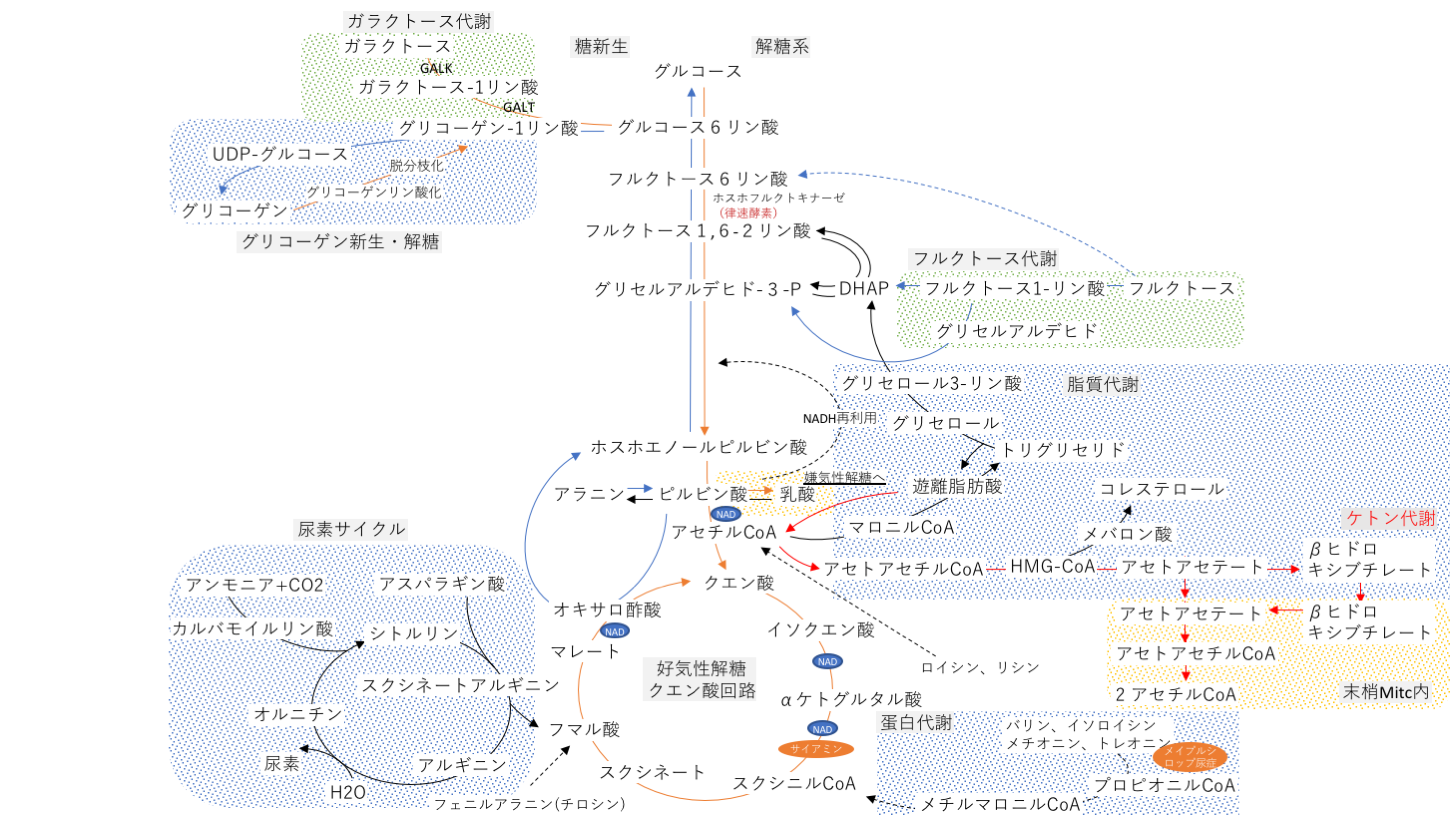
フルクトースはホスホフルクトキナーゼを経ずに代謝されるため、最も分解が早い糖である。
細胞内酵素
ミトコンドリア:脂肪酸のβ酸化、クエン酸回路、ピルビン酸カルボキシル化(糖新生)、ケトン新生に関わる酵素が存在
細胞質ゾル:解糖系、脂肪酸合成、ペントースリン酸経路に関わる酵素が存在
ミトコンドリアと細胞質ゾルの両者:両者の相互作用による、ヘム合成、尿素回路、糖新生に関わる酵素は両者に存在。
クエン酸回路
アセチルCoAからNADHやFADH2やGTPの形でエネルギーが形成されるミトコンドリア内の反応。
- グルコースがピルビン酸に変換される(この過程でNAD+を要するが、これは主にピルビン酸から乳酸への変換時に生じるNAD+から供給される)
- サイアミン依存性のピルビン酸デヒドロゲナーゼによってアセチルCoAに変換される
- クエン酸
- イソクエン酸
- αケトグルタル酸に変換。NADH産生。
- サイアミン依存性のαケトグルタルデヒドロゲナーゼによってスクシニルCoAへ。NADH産生。
- サクシネートへ。GTP産生。
- フマル酸へ。FADH2産生。
- マレート
- オキサロ酢酸へ。NADH産生。
- クエン酸の順に変換される
β酸化
ミトコンドリア内で脂肪酸が酸化されて、FADH2、NADHがATP産生のために、アセチルCoAがクエン酸回路のために作られる。
成人の場合1−2日の絶食によって、小児の場合は8-10時間の絶食でケトン代謝となる。
β酸化の障害はアセチルCoAデヒドロゲナーゼ欠損が最多である。
ミトコンドリアの膜の脂肪酸の通過はカルニチンという膜通過キャリアに依存しており、これはマロニルCoAにより抑制される。
カルニチン欠損症の場合、脂肪酸がミトコンドリアに取り込むことが障害され、脂肪組織から筋組織ではATPが産生できず、肝臓ではケトンが産生できない。低ケトン性低血糖となる。易疲労感。
十分な栄養時 脂肪酸合成
豊富なATPが肝細胞に存在している場合、イソクエン酸デヒドロゲナーゼはATPにより抑制され、ミトコンドリア内のクエン酸濃度は上昇する。クエン酸はミトコンドリア内からシャトルにより細胞質ゾルへ輸送され、アセチルCoAへ変換される。クエン酸の高濃度とインスリン高濃度によって、細胞質ゾルのアセチルCoAカルボキシラーゼは亢進し、マロニルCoAに変換される。マロニルCoAは脂肪酸合成酵素により4炭素分子、16炭素脂肪酸が合成される。
このような場合、マロニルCoAによりβ酸化は、ミトコンドリアへの脂肪酸輸送停止により制限される。
インスリン欠乏時の代謝
1型糖尿病でインスリン欠乏時、DKAとなる。これは脂肪細胞でのトリグリセリドの分解亢進による。
脂肪組織に貯蓄されていたトリグリセリドはホルモン感受性リパーゼにより遊離脂肪酸とグリセロールに分解される。グリセロールは脂肪組織では代謝できないため、血流に入り、肝臓にてグリセロールキナーゼによりグリセロール3リン酸にリン酸化され、デヒドロオキシアセトンリン酸(DHAP)と続いて変換される。DHAPは糖新生もしくは解糖される。
遊離脂肪酸はβ酸化およびケトン形成される。
飢餓時の代謝
飢餓開始12−18時間は肝臓に貯蔵されていたグリコーゲンの分解によりグルコースを産生する。グリコーゲン解糖は肝臓からはグルカゴン、エピネフリンが刺激となりcAMPを介してPhosphorylase kinaseが活性化することで、また骨格筋では主に筋収縮によるCa上昇がトリガーとなって同酵素が活性化して、グリコーゲン解糖が活性化する。
グリコーゲンか枯渇した後は、乳酸、グリセロール、アミノ酸からの糖新生が血中グルコース濃度を保つ手段となる。クエン酸回路のマレイン酸を介して、オキサロ酢酸、ホスホエノールピルビン酸となってグルコースに変換される。オキサロ酢酸からホスホエノールピルビン酸への変換に必要なGTPはクエン酸回路のスクシニルCoAからスクシネートへ変換する際に産生されるGTPから直接供給される。
脂肪細胞において、カテコラミン、グルカゴン、ACTHで促進し、インスリンで抑制されるホルモン感受性リパーゼがある。これにより貯蔵されていたトリグリセリドが、グリセロールと遊離脂肪酸に分解される。これらは共に肝臓に取り込まれ、グリセロールは糖新生へ入り、遊離脂肪酸はケトン体形成される。遊離脂肪酸とケトン体は飢餓時の組織エネルギー源となる。(例外は脳が遊離脂肪酸がBBBを通過できずケトン体のみを使用することと、ミトコンドリアがないため赤血球がグルコースのみを用いること)
遊離脂肪酸は肝臓のミトコンドリア内でケトン(アセトアセテート、βヒドロキシブチル酸)となり、血中に分布。末梢組織のミトコンドリア内で、アセチルCoAに戻る過程でエネルギーを産生する。
糖新生
乳酸、アラニン等はピルビン酸となり、ミトコンドリア内に入り、ビオチン依存性のピルビン酸カルボキシラーゼによりオキサロ酢酸となる。クエン酸回路をマレイン酸にバイパスし、ミトコンドリアを出る。細胞質でマレイン酸からオキサロ酢酸となり、GTPを消費してホスホエノールピルビン酸を介してグルコースが新生される。
ピルビン酸はアセチルCoAに変換されればクエン酸回路に入りエネルギー源となり、オキサロ酢酸となれば糖新生の材料となる。アセチルCoA増加時(脂肪のβ酸化で供給が増えた場合)は、ピルビン酸カルボキシラーゼは促進され糖新生が亢進し、アセチルCoAに変換するピルビン酸デヒドロゲナーゼは抑制されてエネルギー生産が減るといった調節スイッチの機能となっている。
コルチゾールは糖新生に関わる酵素の転写を亢進させ、血糖値を維持する働きがある。
糖新生ではNAD+を用いて反応を進めるが、アルコール飲酒はアルコール分解にNAD+を消費するため、糖新生が抑制され(特に乳酸からピルビン酸への変換が阻害される。)、低血糖となる。
ジサッカロイドの分解
- サッカロース:フルクトース+グルコース
- ラクトース:ガラクトース+グルコース
- マルトース:グルコース+グルコース
解糖系
RBCはミトコンドリアがないため、クエン酸回路を使うことができない。そのため、解糖系を用いてATPを産出している。1,3-BPGをPhosphoglycerate kinaseで3-Phopshoglycerateに変換する際にATPが産生される。また、RBCは1,3-BPGから2,3-BPGを介して3-PhosphoglycerateにATPを得ないで変換することもあり、これは2,3-BPGによる酸素乖離能を用いるためである。低酸素時や慢性貧血時に2,3-BPGが増え、解離曲線の左方移動して、結果として酸素運搬能が増加する。
HbFは2,3-BPGによる影響を受けずに、強い酸素結合を呈する。
グルコースーアラニンサイクル
筋肉で異化したアミノ酸はアラニンとなって肝臓に戻る。肝臓に戻ったアラニンはα-ketoglutarateを用いてピルビン酸となり、グルコースとなる(糖新生)。アラニンからピルビン酸への変換時にはビタミンB6(ピリドキシン)がアミノ酸転基として用いられる。フルクトース2,6-ビスリン酸の高値はアラニンからの糖新生を抑制する。
ピリドキシンはオキサロ酢酸からアスパラギン酸への変換時にも用いられる。
ガラクトース代謝
ガラクトキナーゼ(GALK)欠損症:白内障。
ガラクトース1リン酸ウリディルトランスフェラーゼ(GALT)欠損:ガラクトース1リン酸の蓄積により黄疸、嘔吐、肝腫大、腎障害、大腸菌敗血症、白内障、溶血性貧血を呈する。
メイプルシロップ尿症
常染色体劣性遺伝。分枝アミノ酸(ロイシン、イソロイシン、バリン)のプロピオニルCoAへの分解障害による蓄積症状。(神経毒素=痙攣、イライラ、倦怠感)イソロイシン分解物による甘い香りの尿。分枝アミノ酸分解の補酵素であるチアミン大量投与で改善するケースもあるが、多くは、障害に渡る分枝アミノ酸の食事制限が必要となる。
乳酸デヒドロゲナーゼ欠損
運動時に乳酸合成ができなくなり、解糖系のNAD+が再利用できなくなるため、解糖系が抑制されてしまい、運動時の筋のエネルギー不足となる。
遺伝性フルクトース不耐症
食事より摂取されたフルクトースは肝臓でフルクトキナーゼによりフルクトース1リン酸となり、アルドラーゼBによりジヒドロキシアセトンリン酸とグリセルアルデヒドとなり、解糖系へ入る。
アルドラーゼBが欠損することにより、フルーツ摂取等によりフルクトース1リン酸が蓄積し、嘔吐等の症状、また肝、腎不全を呈する。
フルクトキナーゼのみ欠損時は尿からフルクトースが排泄されるため、予後は良好である(本態性フルクトース尿症)。通常の代謝経路で代謝できないフルクトースはヘキソキナーゼによりフルクトース6リン酸に変換され、ペントースリン酸経路もしくはグリコーゲン合成へ用いられる。
銅還元糖検査で還元糖(グルコース、ガラクトース、フルクトースは)は尿より検出される。
ピルビン酸デヒドロゲナーゼ欠損症
グルコースがピルビン酸に変換された後に、TCAサイクルに入るためにはピルビン酸デヒドロゲナーゼが必要であるが、欠損しているため、乳酸へ代謝される状態。ケトン体による食事(リシンやロイシンはケトン性のアミノ酸)へ変更することで、乳酸産生を抑制することができる。
低酸素状態での代謝
低酸素状態では細胞内にNADHが蓄積するため、ピルビン酸デヒドロゲナーゼが抑制される。このため、ピルビン酸から乳酸への変換が亢進し、重篤な場合は乳酸アシドーシスとなる。
また再灌流時には、還流された酸素により活性酸素種(ROS)が形成される。活性酸素種はDNA変異やタンパク合成阻害の原因となる。抗酸化酵素(superoxide dismutase、グルタチオンペルオキシダーゼ、カタラーゼ)は活性酸素種を酸素と水に分解し中和する働きがある。これらのキャパシティを超えてROSが産生された場合は細胞死につながる。
マックアルディ病
筋肉でのグリコーゲンリン酸化酵素のミオフォスファレースが欠損しているため、筋でのグリコーゲンがグルコース1リン酸に変換できず、筋の糖欠乏症状が運動後早期でる病態。運動前にグルコース摂取することで治療でいる。
コリ病
グリコーゲンの脱分枝酵素の欠損により短鎖の残ったグリコーゲンが蓄積する。肝線維化、筋緊張低下、低血糖、ケトアシドーシス
ポンペ病
グリコーゲンは基本的には細胞質ゲルで分解されるが、肝や心筋細胞では少量リソソームにグリコーゲンが紛れ込む。これがアシッドαグルコシダーゼの欠損により、グリコーゲンが蓄積することで、リソソームが膨張し、細胞の機能障害となる。PAS染色で陽性のグリコーゲンがリソソーム内に認める。
ハートナップ病
常染色体劣性遺伝。中性アミノ酸の輸送障害で特にトリプトファンが障害される。トリプトファンはナイアシンの前駆体であり、ナイアシン欠乏症状を呈する。ペラグラ様皮疹、小脳失調。
遺伝性オロチン酸尿症
常染色体劣性遺伝。大球性貧血、発達障害。ピリミジン合成の過程において、オロチン酸からウリジン5−モノリン酸(UMP)変換酵素が欠損のため、UMPが産生できない。ウリジンを補充することで、UMPを合成し、症状を改善できる。
尿素サイクル
アミノ酸から生じたアンモニアを尿素に変換するサイクル。
ミトコンドリアに取り込まれたオルニチンと、アンモニアから生成されたカルバモイルリン酸をオルニチントランスカルバミラーゼを用いてシトルリンに変換する。ミトコンドリア外でシトルリンはアルギニンを経て、尿素とオルニチンに変換される。
・ミトコンドリア内へのオルニチン輸送の障害のある児では、蛋白制限が必要となる。
・サイクル内でアルギニンを尿素に変換するアルギナーゼ欠損の児ではアルギニン蓄積をきたす。症状は四肢痙性麻痺、舞踏様運動
・オルニチントランスカルバミラーゼ欠損症では、ストレス時にアンモニアが蓄積し、嘔吐、意識障害が生じる。
ペントースリン酸経路
G6PDからリブロース5リン酸に代謝する過程でNADPHを産生する。その後リブロース5リン酸はフルクトース6リン酸となり、解糖系へ入る。
この過程で産生されるNADPHにより、抗酸化物質であるグルタチオンを産生され、また、脂肪酸やコレステロール、ステロイドの産生やチトクロームP450の代謝のためにNADPHは用いられる。
カテコラミン合成、チロシン代謝
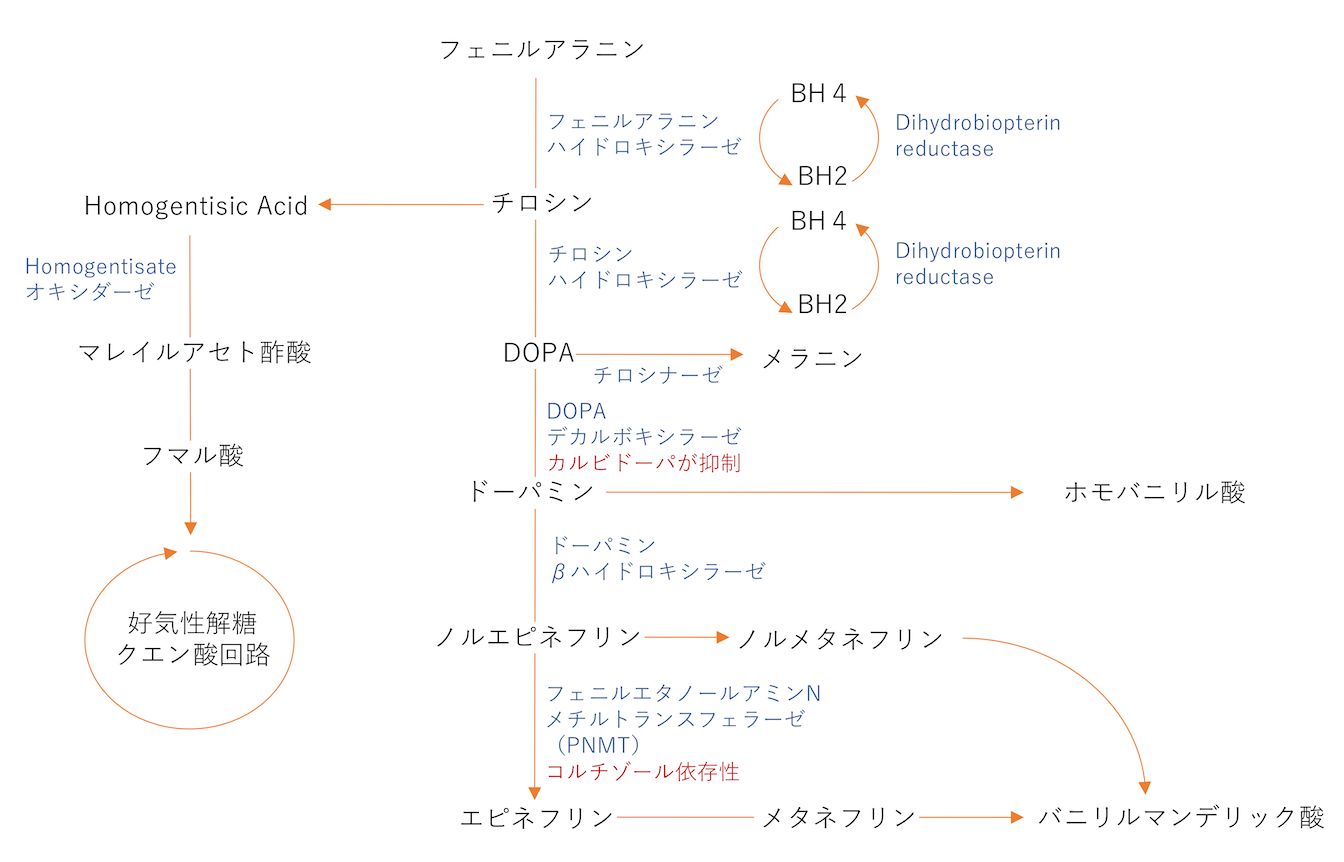
PNMTはコルチゾール依存性であり、コルチゾールが欠損するとエピネフリンの合成が欠乏する。
フェニルケトン尿症 PKU
フェニルアラニンからDOPAとメラニンに変換する過程のPhenylalanine hydroxylase(98%はこのタイプ)もしくはTetrahydrobiopterin(BH4)の低値による。この2つはTryptophanからセロトニンを作る際にも必須であり、フェニルケトン尿症患者はセロトニン欠損にもなる。
フェニルケトンからチロシンが産生できないため、チロシン補充が必須となる(多くの患者は食事から十分量補充できる。)。
カビ臭い匂い(musty odor)、神経発達障害、痙攣、色素欠損。治療はフェニルアラニンの摂取制限。
メラニンの欠乏により、皮膚、髪、目およびカテコラミン作動性脳神経核の色素欠損となる。
BH4低値のタイプはほとんどがBH2をBH4に変換する酵素(Dihydrobiopterin reductase)の欠損による。この場合は、チロシンからDOPAの変換もできないため、DOPAやその下流のドーパミン、エピネフリン、ノルエピネフリン、セロトニンも欠乏し、神経の発達障害となる。ドーパミン低値のためプロラクチンの分泌が亢進する。
アルカプトン尿症
homogenitisic acid dioxygenaseの欠損によるもの。homogentisic acidの蓄積は結合組織の色素沈着となり、青黒い沈着が角膜や耳軟骨に認められる。大関節や脊椎にも沈着を認め、脊椎強直や痛み、可動制限になりうる。尿中に排出されたものは、空気中で酸化されてアルカプトンとなり黒色尿となる。
Propionyl-CoAとなるもの
Valine, Isoleucine, Methionine, Threonineといった必須アミノ酸、Odd-chain fatty acidsが材料となる。
これらはPropionyl-CoAを経てSuccinyl-CoAとなり、TCAサイクルに入る。